NORMA Oficial Mexicana NOM-137-SSA1-2025, Etiquetado de dispositivos médicos.
Al margen un sello con el Escudo Nacional, que dice: Estados Unidos Mexicanos.- Salud.- Secretaría de Salud.- Comisión Federal para la Protección contra Riesgos Sanitarios.
VICTOR HUGO BORJA ABURTO, Comisionado Federal para la Protección contra Riesgos Sanitarios y Presidente del Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario con fundamento en los artículos 39 de la Ley Orgánica de la Administración Pública Federal; 4 de la Ley Federal de Procedimiento Administrativo; 2, fracciones II y IV, 3o, fracciones XXIII y XXIV, 17 bis, fracciones II, III y VI, 194, fracción II, 194 bis, 195, 201, 210, 212, 213, 214, 262, 263, 264, 265, 266 y 376 de la Ley General de Salud; 3, fracción IX, 10, fracción I, 30, 34, 35, 37, 38 y 139 de la Ley de Infraestructura de la Calidad; 28 y 33 del Reglamento de la Ley Federal sobre Metrología y Normalización en relación con el Transitorio Tercero del Decreto por el que se expide la Ley de Infraestructura de la Calidad y se abroga la Ley Federal sobre Metrología y Normalización; 2o, fracción VIII, 7o, fracción IV, 8, primer párrafo, 11, 20, 23, 24, 165, 183, fracción III del Reglamento de Insumos para la Salud; 3, fracciones I, literal b y II, así como 10, fracciones IV y VIII del Reglamento de la Comisión Federal para la Protección contra Riesgos Sanitarios, y
CONSIDERANDO
Que la Norma Oficial Mexicana NOM-137-SSA1-2008, Etiquetado de dispositivos médicos fue publicada en el Diario Oficial de la Federación el 12 de diciembre de 2008, con el objeto de establecer los requisitos mínimos para comunicar la información a los usuarios, que deberá contener el etiquetado de los dispositivos médicos (equipo médico, prótesis, órtesis, ayudas funcionales, agentes de diagnóstico, insumos de uso odontológico, materiales quirúrgicos, de curación y productos higiénicos) de origen nacional o extranjero, que se comercialicen o destinen a usuarios en el territorio nacional;
Que esta Norma fue inscrita por primera vez en el Programa Nacional de Infraestructura de la Calidad 2021, siendo incluida subsecuentemente en los programas de los años siguientes, inclusive en el Programa de 2025, con el objeto de ser modificada a fin de establecer controles y procedimientos más precisos respecto de los requisitos mínimos que deberá contener el etiquetado de los dispositivos médicos, que sirvan para comunicar la información a los usuarios;
Que con fecha 23 de abril de 2024, en cumplimiento del acuerdo del Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario y de lo previsto por el artículo 35, fracción V de la Ley de Infraestructura de la Calidad, se publicó en el Diario Oficial de la Federación, el Proyecto de la presente Norma, a efecto de que dentro de los 60 días naturales siguientes a dicha publicación, los interesados presentaran sus comentarios ante dicho Comité;
Que con fecha previa, fue publicada en el Diario Oficial de la Federación, la respuesta a los comentarios recibidos por el mencionado Comité, en los términos del artículo 47, fracción VI, de la Ley de Infraestructura de la Calidad, y
Que en atención a las anteriores consideraciones, contando con la aprobación del Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario, he tenido a bien expedir y ordenar la publicación en el Diario Oficial de la Federación de la
NORMA OFICIAL MEXICANA NOM-137-SSA1-2025, ETIQUETADO DE DISPOSITIVOS MÉDICOS
PREFACIO
En la elaboración de la presente Norma Oficial Mexicana participaron:
SECRETARÍA DE SALUD.
Comisión Federal para la Protección contra Riesgos Sanitarios.
Comisión Permanente de la Farmacopea de los Estados Unidos Mexicanos.
Dirección General de Modernización del Sector Salud.
CONSEJO DE SALUBRIDAD GENERAL.
INSTITUTO MEXICANO DEL SEGURO SOCIAL.
Coordinación de Calidad de Insumos y Laboratorios Especializados.
INSTITUTO DE SEGURIDAD Y SERVICIOS SOCIALES DE LOS TRABAJADORES DEL ESTADO.
Subdirección de Infraestructura.
UNIVERSIDAD NACIONAL AUTÓNOMA DE MÉXICO.
Facultad de Química.
INSTITUTO POLITÉCNICO NACIONAL.
Escuela Nacional de Ciencias Biológicas.
UNIVERSIDAD AUTÓNOMA METROPOLITANA.
Iztapalapa-División de Ciencias Básicas e Ingeniería.
CÁMARA NACIONAL DE LA INDUSTRIA DE TRANSFORMACIÓN.
Sector Industrial Médico.
CÁMARA NACIONAL DE LA INDUSTRIA FARMACÉUTICA.
Sección de Productos Auxiliares para la Salud.
Sección de Reactivos y Sistemas de Diagnóstico.
ACADEMIA NACIONAL DE CIENCIAS FARMACÉUTICAS, A.C.
ASOCIACIÓN FARMACÉUTICA MEXICANA, A.C.
COLEGIO NACIONAL DE QUÍMICOS FARMACÉUTICOS BIÓLOGOS MÉXICO, A.C.
ASOCIACIÓN MEXICANA DE LABORATORIOS FARMACÉUTICOS, A.C.
ASOCIACIÓN MEXICANA DE INDUSTRIAS INNOVADORAS DE DISPOSITIVOS MÉDICOS, A.C.
COLEGIO DE INGENIEROS BIOMÉDICOS DE MÉXICO, A.C.
ÍNDICE
0. Introducción
1. Objetivo y campo de aplicación
2. Referencias normativas
3. Términos y definiciones
4. Símbolos y términos abreviados
5. Requisitos generales
6. Requisitos específicos para el etiquetado de los dispositivos médicos
7. Concordancia con Normas Internacionales
8. Bibliografía
9. Observancia de la Norma
10. Evaluación de la conformidad
11. Vigencia
Apéndice A (Normativo) Símbolos para el etiquetado de los dispositivos médicos
0. Introducción
Los dispositivos médicos se consideran como parte fundamental de los sistemas de salud y los beneficios que proporcionan continúan aumentando día con día, ya que son esenciales para prevenir, diagnosticar, tratar y rehabilitar enfermedades de una manera segura y efectiva.
Por otro lado, los dispositivos médicos funcionan como reanimador y paliativo, así como complemento integro de estrategias inter-sectoriales para una cobertura universal de salud; en el diagnóstico de brotes y durante emergencias, mejoran el bienestar del paciente, siendo necesarios para realizar intervenciones en los diferentes niveles de atención médica.
Estos pueden variar desde un apósito simple a un abatelenguas, a un kit de pruebas clínicas, a una cadera de reemplazo, hasta para diagnosticar SARS-CoV-2, Cáncer, el VIH/SIDA; o como coadyuvante en el tratamiento de enfermedades cardiovasculares y diabetes, así como otros dispositivos que son utilizados para sustituir, modificar o apoyar la anatomía o a un proceso fisiológico, reproductivo, materno, neonatal o infantil, siendo en algunos casos de vital importancia para la salud del ser humano.
Las tecnologías sanitarias son esenciales para que el sistema de salud funcione, y los dispositivos médicos en particular, son cruciales en la prevención, el diagnóstico y el tratamiento de enfermedades y dolencias, así como en la rehabilitación de los pacientes, y estos son utilizados por personal paramédico, médicos en clínicas remotas, por optometristas y ópticos, oftalmólogos, dentistas y por otros profesionales de la salud en instalaciones médicas avanzadas con fines médicos.
Por lo anterior, los dispositivos médicos deben incluir en su envase o empaque primario o empaque secundario un etiquetado visible con información sanitaria que se relacione con la identificación del dispositivo médico, la identidad del fabricante, la descripción técnica, la finalidad prevista, el uso correcto o el uso previsto, como se debe mantener y almacenar, comunicar cualquier riesgo residual, advertencia, limitación o contraindicación que deba tomarse para apoyar y asistir a los usuarios del dispositivo médico en su uso seguro y apropiado.
El etiquetado del dispositivo médico debe contener información general y específica, proporcionada por el fabricante con el uso previsto para informar al usuario, paciente, profesional de la salud y población en general de las características del dispositivo médico de cualquier contraindicación, advertencia o precaución que deba considerarse para apoyar y asistir a los usuarios del dispositivo en su uso seguro y desempeño y así poder evitar incidentes adversos.
1. Objetivo y campo de aplicación
1.1 Objetivo
La presente Norma Oficial Mexicana tiene por objeto establecer los requisitos de información sanitaria que debe contener el etiquetado de los dispositivos médicos para uso humano, el uso correcto y trazabilidad de los mismos, que se destinen a usuarios o se comercialicen y que se pongan a disposición en territorio nacional.
1.2 Campo de aplicación
La presente Norma Oficial Mexicana es de observancia obligatoria en todo el territorio nacional para los establecimientos que se dediquen a la fabricación, acondicionamiento, distribución o importación de dispositivos médicos con fines de comercialización o suministro en México.
2. Referencias normativas
Para la correcta aplicación de esta Norma, es necesario consultar las siguientes Normas:
2.1 Norma Oficial Mexicana NOM-008-SE-2021, Sistema general de unidades de medida (cancela a la NOM-008-SCFI-2002).
2.2 Norma Oficial Mexicana NOM-050-SCFI-2004, Información comercial-Etiquetado general de productos.
2.3 Norma Oficial Mexicana NOM-072-SSA1-2012, Etiquetado de medicamentos y remedios herbolarios.
2.4 Norma Oficial Mexicana NOM-240-SSA1-2012, Instalación y operación de la tecnovigilancia.
2.5 Norma Oficial Mexicana NOM-241-SSA1-2025, Buenas prácticas de fabricación de dispositivos médicos.
2.6 Farmacopea de los Estados Unidos Mexicanos, Suplemento para dispositivos médicos 5.0.
2.7 Farmacopea de los Estados Unidos Mexicanos, Suplemento para Establecimientos dedicados a la venta y suministro de medicamentos y demás Insumos para la Salud, 6a ed.
3. Términos y definiciones
Para efectos de esta Norma Oficial Mexicana se entiende por:
3.1 Accesorio de un dispositivo médico, al artículo que, sin ser en sí mismo un dispositivo médico, está destinado por su fabricante a ser usado en forma conjunta con uno o varios de dichos productos, para permitir específicamente que el producto pueda utilizarse para su indicación de uso o para contribuir específica y directamente a la funcionalidad médica de los dispositivos médicos.
Estos pueden venir incluidos dentro del mismo empaque del dispositivo médico, o bien pueden suministrarse por separado como un accesorio para uso exclusivo con el dispositivo médico principal.
3.2 Acondicionamiento, a todas las operaciones a las que tiene que someterse un producto a granel hasta llevarlo a su presentación como producto terminado.
3.3 Advertencia, a la declaración que alerta a los usuarios acerca de una situación que, cuando no se evita, podría dar lugar a peligros u otras consecuencias adversas graves a partir de la utilización de un dispositivo médico.
3.4 Agente de diagnóstico, a todos los insumos incluyendo antígenos, anticuerpos, calibradores, verificadores, reactivos, equipos de reactivos, medios de cultivo y de contraste y cualquier otro similar que pueda utilizarse como auxiliar de otros procedimientos clínicos o paraclínicos.
NOTA: los agentes de diagnóstico son dispositivos médicos utilizados solos o en combinación con otros dispositivos médicos, destinados a proporcionar información para la detección, pronóstico, diagnóstico o monitoreo de condiciones fisiológicas, estados de salud, enfermedades o malformaciones congénitas en humanos, a partir del examen in vitro de muestras derivadas del cuerpo humano y/o por la administración de medios de contraste y radiofármacos.
3.5 Agente de diagnóstico in vitro (DIV o IVD, por sus siglas en inglés), al dispositivo médico utilizado solo o en combinación, destinado por el fabricante para el examen in vitro de muestras derivadas del cuerpo humano, única o principalmente para proveer información para el diagnóstico, monitoreo o compatibilidad.
NOTA 1: reactivos, calibradores, dispositivos de recolección y almacenamiento de muestras, materiales de control e instrumentos, aparatos o productos relacionados.
NOTA 2: puede usarse solo o en combinación con accesorios u otros dispositivos médicos.
3.6 Agentes de diagnóstico in vitro para autopruebas, al dispositivo médico para diagnóstico in vitro que tiene la finalidad de ser utilizado por personal que no cuenta con un entrenamiento formal para su uso.
3.7 Almacenamiento, a la conservación de insumos, producto a granel, semiterminado y terminado del dispositivo médico, que se conservan en un área con condiciones establecidas de acuerdo con su nivel de riesgo.
3.8 Calibración, a la demostración de que un instrumento particular o dispositivo médico produce resultados dentro de la especificación, en comparación con los producidos por una referencia o estándar trazable sobre un intervalo de medición establecido.
3.9 Componente, a cualquier sustancia, material o ingrediente utilizado en la fabricación de un dispositivo médico presente en el producto final.
3.10 Condiciones de almacenamiento, a las requeridas para preservar o conservar las características de calidad de los insumos, producto a granel, semiterminado y terminado.
3.11 Contraetiqueta, a la etiqueta adicional que contiene la información sanitaria complementaria, cuando la etiqueta de origen no cumple parcial o totalmente con la presente Norma.
3.12 Contraetiquetado, a la actividad de colocar contraetiquetas al dispositivo médico terminado posterior al envasado primario o secundario.
3.13 Contraindicación, a los elementos del etiquetado que describen situaciones como los grupos de pacientes, las razones médicas o las afecciones clínicas en las cuales el dispositivo médico no debe usarse porque el riesgo de utilización es claramente superior a todo beneficio posible.
3.14 Consumibles, a los materiales necesarios para que el dispositivo médico realice sus funciones, desechables, que con su operación se agotan y que son de consumo repetitivo.
3.15 Denominación distintiva, al nombre que, como marca comercial, le asigna el laboratorio o fabricante a sus dispositivos médicos con el fin de distinguirlos de otros similares.
3.16 Denominación genérica, al nombre que describe a un dispositivo médico o grupo de dispositivos médicos que tienen características comunes, propuesto por el fabricante o el Titular del Registro Sanitario, reconocido internacionalmente y aceptado por la autoridad sanitaria.
3.17 Desempeño analítico de un agente de diagnóstico in vitro, a su capacidad para detectar o medir correctamente un analito concreto.
3.18 Desempeño clínico de un agente de diagnóstico in vitro, a su capacidad para producir resultados que se correlacionan con una condición clínica o con un proceso, o estado fisiológico o patológico específico, en función de la población destinada y del usuario previsto.
3.19 Desempeño clínico del dispositivo médico, a la capacidad resultante de todos los efectos médicos directos o indirectos derivados de sus características técnicas o funcionales, para alcanzar la indicación de uso, generando así beneficios clínicos para los pacientes, cuando se utilice para lograr su propósito previsto según lo declarado por el fabricante.
3.20 Desempeño de un agente de diagnóstico in vitro, a su capacidad de lograr su uso o finalidad prevista, indicada por el fabricante. El desempeño de un agente de diagnóstico in vitro está compuesto por el funcionamiento analítico y, en su caso, el funcionamiento clínico que contribuya a la finalidad prevista del agente de diagnóstico in vitro.
3.21 Desempeño de un dispositivo médico, a la capacidad de un dispositivo médico para lograr su uso previsto conforme a lo establecido por el fabricante. El desempeño puede incluir aspectos tanto clínicos como técnicos.
3.22 Dispositivo médico de un solo uso, al destinado a utilizarse en un usuario determinado, durante o para un procedimiento único y luego se desecha. No está previsto su reprocesamiento para usarlo nuevamente, de acuerdo con las indicaciones de uso del fabricante.
3.23 Dispositivo médico, al instrumento, aparato, utensilio, máquina, software, producto o material implantable, agente de diagnóstico, material, sustancia o producto similar, para ser empleado, solo o en combinación, directa o indirectamente en seres humanos; con alguna(s) de las siguientes finalidades de uso:
- Diagnóstico, prevención, vigilancia o monitoreo, y/o auxiliar en el tratamiento de enfermedades;
- Diagnóstico, vigilancia o monitoreo, tratamiento, protección, absorción, drenaje, o auxiliar en la cicatrización de una lesión;
- Sustitución, modificación o apoyo de la anatomía o de un proceso fisiológico;
- Soporte de vida;
- Control de la concepción;
- Desinfección de dispositivos médicos;
- Sustancias desinfectantes;
- Provisión de información mediante un examen in vitro de muestras extraídas del cuerpo humano, con fines diagnósticos;
- Dispositivos que incorporan tejidos de origen animal y/o humano, y/o
- Dispositivos empleados en fertilización in vitro y tecnologías de reproducción asistida.
Y cuya finalidad de uso principal no es a través de mecanismos farmacológicos, inmunológicos o metabólicos, sin embargo, pueden ser asistidos por estos medios para lograr su función. Los dispositivos médicos incluyen a los Insumos para la salud de las siguientes categorías: equipo médico, prótesis, órtesis y ayudas funcionales, agentes de diagnóstico, insumos de uso odontológico, materiales quirúrgicos, de curación, productos higiénicos y los demás insumos que sean considerados para este uso y sean evaluados y reconocidos como dispositivos médicos por la Secretaría de Salud a solicitud.
3.24 Dispositivos médicos que incorporan un fármaco o medicamento, a aquellos que incluyen como parte integral un fármaco o medicamento que ejerce sobre el cuerpo humano una acción secundaria o adicional a la del dispositivo médico.
3.25 Distribuidor, a la persona física o moral que almacena o distribuye, o ambas, y en su caso importa insumos, y que cuenta con aviso de funcionamiento o licencia sanitaria dependiendo del giro de productos que comercialice.
3.26 Envase o empaque primario, a los elementos del sistema de envase que estén en contacto directo con el dispositivo médico.
3.27 Envase o empaque secundario, a los elementos que forman parte del empaque en el cual se comercializa el dispositivo médico y que no están en contacto directo con él.
3.28 Envase o empaque múltiple o colectivo, a cualquier recipiente o envoltura en el que se encuentran contenidos dos o más envases o empaques primarios o secundarios.
3.29 Estabilidad, a la capacidad de un dispositivo médico de permanecer dentro de las especificaciones de calidad establecidas, en el envase o empaque primario que lo contiene, o secundario, cuando éste sea una condición esencial para su vida útil esperada.
NOTA 1: estabilidad aplica a:
· dispositivos médicos estériles y no estériles en los que las propiedades físicas, químicas o funcionales puedan verse alteradas o afectadas en un lapso declarado;
· reactivos de diagnóstico in vitro, calibradores y controles, cuando se almacenen, transporten y utilicen en las condiciones especificadas por el fabricante;
· materiales liofilizados reconstituidos, soluciones de trabajo y material extraído de envases sellados (cuando se preparen, usen y almacenen conforme a las instrucciones de uso del fabricante).
NOTA 2: la estabilidad de un reactivo o un sistema de medición de diagnóstico in vitro se suele cuantificar con respecto al tiempo y en condiciones definidas:
· en términos de la duración de un intervalo de tiempo durante el cual se modifica una propiedad medida en una magnitud declarada;
· en términos del cambio de una propiedad en un intervalo de tiempo determinado.
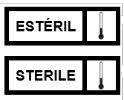
3.30 Estéril, a la ausencia de microorganismos viables y esporas.
3.31 Esterilización, al proceso validado utilizado para dejar un producto libre de todos los microorganismos viables, incluidas las esporas bacterianas.
3.32 Etiqueta, al marbete, rótulo, marca o imagen gráfica que se haya escrito, impreso, estarcido, marcado, marcado en relieve o en hueco, grabado, adherido o precintado en cualquier material susceptible de contener el dispositivo médico, incluyendo el envase mismo.
3.33 Etiquetado, al conjunto de elementos que incluyen la etiqueta, contraetiqueta, manual, instrucciones de uso y cualquier otra información suministrada por el fabricante o por el titular del registro sanitario que se relacione con la identificación, la descripción técnica, la indicación de uso y el uso correcto del dispositivo médico, con exclusión de los documentos de envío.
3.34 Etiquetado electrónico, a cualquier forma de contenido de etiquetado suministrada en un medio electrónico accesible, por el fabricante en relación con un dispositivo médico.
3.35 Fabricación, a las operaciones involucradas en la producción y acondicionamiento de un dispositivo médico, desde la recepción de insumos, liberación, almacenamiento y distribución como producto terminado.
3.36 Fabricante, a la persona física o moral, responsable de la fabricación, rehabilitación, remanufactura, ensamblado de un sistema, acondicionamiento, etiquetado, liberación como producto terminado; de la calidad de un dispositivo médico terminado previo a su comercialización bajo su propio nombre.
NOTA: cuando no se declare un fabricante legal se entenderá que esta figura, es responsable del diseño, seguridad y desempeño.
3.37 Fabricante legal, a la persona física o moral, responsable de la seguridad, desempeño, calidad; diseño, fabricación, rehabilitación, remanufactura, ensamblado de un sistema; acondicionamiento, etiquetado, liberación como producto terminado de un dispositivo médico, por esta misma persona, independientemente de que estas operaciones sean efectuadas por un tercero.
NOTA 1: se consideran fabricantes legales, a aquellas compañías comercializadoras que adquieren el dispositivo médico terminado a un OEM (Original Equipment Manufacturer) y utilizan los contratos de manufactura original de equipo con su marca propia para productos manufacturados totalmente por otros fabricantes.
NOTA 2: el fabricante legal puede comercializar bajo su propio nombre.
3.38 Fecha de caducidad, a la que indica el fin del periodo de vida útil esperada del dispositivo médico y está basado en los estudios de estabilidad.
3.39 Fecha de fabricación o elaboración, a la fecha en la que se fabricó o elaboró el dispositivo médico.
3.40 Gestión de riesgos, a la aplicación sistemática de las políticas de gestión, los procedimientos y las prácticas, a las tareas de análisis, evaluación, control y seguimiento del riesgo.
3.41 Incidente/evento, a cualquier acontecimiento atribuido principalmente al dispositivo médico por su incapacidad para funcionar de acuerdo con su finalidad prevista cuando se utiliza conforme a las instrucciones de uso del fabricante o deterioro de las características del dispositivo médico o cualquier insuficiencia de la información facilitada por el fabricante y que puede o no desencadenar un incidente adverso.
3.42 Incidente adverso (IA)/evento adverso, al acontecimiento asociado por el uso de un dispositivo médico que ha causado o que podría causar un deterioro grave en la salud del usuario e incluso la muerte y que cuenta con pruebas suficientes de la relación causal, comprobada por el Titular del Registro Sanitario o su Representante Legal en México.
3.43 Indicación de uso, a los fines a los que se destina un dispositivo médico según los datos facilitados por el fabricante en el etiquetado, en las instrucciones de uso, según lo indicado por el fabricante en la evaluación clínica, debe señalar el grupo de pacientes destinatarios, afecciones que se pretende diagnosticar, prevenir, tratar o controlar.
3.44 Información de seguridad, a la información proporcionada al usuario u organización responsable que se utiliza como medida de control de riesgos o divulgación de un riesgo residual.
NOTA: los ejemplos pueden incluir advertencias o precauciones, instrucciones sobre el uso de un dispositivo médico para evitar errores de uso o evitar una situación peligrosa, o explicación de una característica de seguridad de un dispositivo médico.
3.45 Instrucciones de uso, a la información general y técnica proporcionada por el fabricante para informar al usuario de un dispositivo médico sobre su indicación de uso, así como cualquier contraindicación, advertencia o precaución que deban tomar, con el fin de apoyar y ayudar a los usuarios en su utilización segura y correcta.
NOTA: las instrucciones de uso también pueden denominarse "instructivo o inserto", y pueden ser suministrados de manera electrónica.
3.46 Juego/Paquete (Kit), a la presentación de dos o más tipos de dispositivos médicos empacados juntos, destinados a utilizarse en la misma determinación o en el mismo procedimiento médico. La clasificación del kit se basa en la clase más alta del dispositivo que se proporciona en el paquete. Deben identificarse todos los componentes del kit en su etiqueta. Cada componente debe cumplir con la normatividad vigente que corresponda.
3.47 Lote, a la cantidad específica de cualquier materia prima, insumo o dispositivo médico, que haya sido elaborado en un ciclo de producción, bajo condiciones equivalentes de operación y durante un periodo determinado.
3.48 Manual, al documento impreso o en formato electrónico que, en forma escrita, gráfica o ambas, acompaña a un dispositivo médico, que contiene información importante para el usuario o aquellos responsables de su instalación, utilización, mantenimiento, desinstalación y la disposición final del dispositivo médico, particularmente en relación con el uso seguro.
3.49 Maquila, al proceso o etapa de un proceso involucrado en la fabricación de un dispositivo médico, realizado por un establecimiento diferente del titular del registro sanitario o fabricante; puede ser nacional o internacional, temporal o permanente.
3.50 Modelo, al nombre o número utilizado para representar un dispositivo médico o una familia de dispositivos médicos que tienen características comunes.
3.51 Muestras sin fines de lucro, al dispositivo médico sin fines de comercialización.
3.52 Número de catálogo, al valor dado por el fabricante para identificar el dispositivo médico específico en lo que se refiere a su forma y ajustes, funcionamiento y procesos (tales como, procesos de fabricación que requieren diferenciación para el usuario final).
3.53 Número de lote o de serie, a la combinación numérica o alfanumérica que identifica específicamente un lote o serie.
3.54 Paciente, a la persona que recibe asistencia de un prestador de atención de salud que puede beneficiarse con la acción de un dispositivo médico. Un paciente también puede ser usuario de un dispositivo médico.
3.55 Población en general, al conjuntó de seres humanos que viven en un determinado espacio geográfico o territorio.
3.56 Precaución, a la leyenda o instrucción que se coloca en un dispositivo médico con el fin de evitar al usuario un daño o peligro en el uso del producto.
3.57 Profesionales de la salud, a las personas con título profesional o certificado de especialización y cédula profesional legalmente expedidos y registrado por las autoridades educativas competentes en relación con las ciencias químico-farmacéuticas, ciencias de la salud humana o áreas afines.
3.58 Prueba de autocomprobación, al uso de un dispositivo médico por parte de un usuario no especializado que es responsable de recopilar los datos o la muestra por sí mismo, basándose únicamente en las instrucciones proporcionadas por el fabricante. Este uso también puede incluir realizar la prueba e interpretar los resultados por sí mismo.
NOTA: la prueba de autocomprobación puede incluir un cuidador externo.
3.59 Pruebas de diagnóstico ambulatorias, a las pruebas realizadas fuera de un laboratorio por un profesional de la salud, que no necesariamente es un profesional laboratorista, generalmente se realizan cerca o en presencia del paciente.
3.60 Prueba rápida, a todas aquellas que se utilizan con la finalidad de escrutinio en las mediciones de los componentes de interés médico en muestras de tejidos, fluidos, excreciones y secreciones del cuerpo humano que dan un resultado rápido, el cual deberá ser confirmado por pruebas de laboratorio y/o clínicamente.
3.61 Reutilizable, a los insumos o dispositivos médicos que pueden ser utilizados en varias ocasiones, ya sea en el mismo o diferentes pacientes. Pueden requerir una descontaminación adecuada y otros reprocesamientos entre usos.
3.62 Riesgo, a la combinación de la probabilidad de ocurrencia de un daño y la gravedad de tal daño.
3.63 Riesgo residual, al riesgo remanente después de que se han tomado las medidas de control del riesgo.
3.64 Seguridad, a la valoración del beneficio que produce un dispositivo médico frente a sus posibles riesgos en un momento dado.
3.65 Señal de seguridad, al rótulo que trasmite un mensaje general de seguridad, obtenido mediante la combinación de un color y una forma geométrica.
3.66 Símbolo, a la representación gráfica que aparece en la etiqueta o la documentación asociada, o ambas, de un dispositivo médico que comunica información característica sin necesidad de que el proveedor o receptor de la información maneje el lenguaje de un país o población en particular.
NOTA: el símbolo puede ser una representación pictórica o gráfica abstracta, o una que utilice objetos familiares, incluidos caracteres alfanuméricos.
3.67 Software como Dispositivo Médico (ScDM), al software utilizado con uno o más propósitos médicos, que tiene como característica principal que no requiere formar parte del hardware del dispositivo médico para cumplir con el propósito médico previsto; es capaz de funcionar en plataformas computacionales generales y puede utilizarse solo o en combinación con otros productos (tales como, módulo, otros dispositivos médicos, etcétera). Las aplicaciones móviles que cumplen con esta definición son consideradas software como dispositivo médico. El software que hace funcionar al dispositivo médico físico queda excluido de esta definición.
NOTA: el ScDM también puede proporcionar medios y sugerencias para mitigar una enfermedad; proporcionar información para determinar la compatibilidad, detección, diagnóstico, monitoreo o tratamiento de condiciones fisiológicas, estados de salud, enfermedades o deformidades congénitas; ser un auxiliar en el diagnóstico, detección, monitoreo, determinación de la predisposición; pronóstico, predicción, determinación del estado fisiológico.
3.68 Suministro, a la disposición, provisión de un dispositivo médico, a través de un mecanismo de comercialización o como muestra sin fines de lucro, con el objetivo de su distribución, utilización o ambas en el mercado.
3.69 Titular del Registro Sanitario, a la persona física o moral que obtiene la autorización otorgada por la Secretaría de Salud, a través de la Comisión Federal para la Protección contra Riesgos Sanitarios.
3.70 Uso previsto/finalidad de uso, al propósito al que está destinado un dispositivo médico, proceso o servicio de conformidad con las especificaciones, las instrucciones e información proporcionada por el fabricante.
NOTA: el uso previsto puede incluir la indicación de uso.
3.71 Usuario(s), a pacientes, profesionales de la salud y población en general que utiliza un dispositivo médico.
3.72 Vida útil esperada/Vida útil prevista, al período de tiempo especificado por el fabricante durante el cual se prevé que se mantenga el uso seguro y efectivo del dispositivo médico.
NOTA 1: la vida útil esperada puede determinarse mediante la estabilidad o mediante otros métodos.
NOTA 2: es posible que sea necesario realizar el mantenimiento, las reparaciones o sus actualizaciones.
4. Símbolos y términos abreviados
Para los propósitos de la presente Norma, se aplican los símbolos y términos abreviados siguientes:
| 4.1 Símbolos |
| 4.1.1 | % | Por ciento |
| 4.1.2 | pH | Potencial de hidrógeno |
| 4.2 | Términos abreviados |
| 4.2.1 | COFEPRIS | Comisión Federal para la Protección contra Riesgos Sanitarios |
| 4.2.2 | RFID | Identificación por radiofrecuencia (por sus siglas en inglés Radio Frequency Identification) |
| 4.2.3 | QR | Respuesta rápida (por sus siglas en inglés Quick Response) |
| 4.2.4 | EDTA | Ácido etilendiaminotetraacético (por sus siglas en inglés ethylenediaminetetraacetic acid) |
| 4.2.5 | OEM | Fabricante de equipamiento original (por sus siglas en inglés original equipment manufacturer) |
| 4.2.6 | ScDM | Software como dispositivo médico |
| 4.2.7 | CMR | cancerígenas, mutagénicas y reprotóxicas |
| 4.2.8 | NEQ | No equivalente |
| 4.2.9 | SGC | Sistema de Gestión de Calidad |
| 4.2.10 | DIV o IVD | Agente de diagnóstico in vitro (por sus siglas en inglés) |
5. Requisitos generales
5.1 Los datos que ostenten las etiquetas o contraetiquetas de los productos objeto de esta Norma, en su envase o empaque de venta (primario, secundario, múltiple o colectivo), de manera enunciativa, más no limitativa, deberán cumplir con lo dispuesto por los artículos 2, fracciones III y IV, 17 bis, fracciones III y VII, 194, fracción II, 194 bis, 195, 210, 212, 214, 263, 264, 265 y 266 de la Ley General de Salud; 2, fracción VIII, 8, primer párrafo, 11, 24, 165, 179, fracciones II, III y IX, 182, 183, fracción III y 184, segundo párrafo del Reglamento de Insumos para la Salud, así como por cualquier disposición legal aplicable a la materia y se expresarán en idioma español en su contenido en términos comprensibles, tipografía y tamaño legibles para el usuario, sin perjuicio de que además se expresen en otros idiomas u otro sistema de medida.
5.2 Toda información sanitaria que se relaciona con la identificación, la descripción técnica, la indicación de uso y el uso correcto debe estar contenida en el etiquetado de los dispositivos médicos objeto de la presente Norma, puede complementarse de manera electrónica a través de identificación por radiofrecuencia (RFID), o códigos de barras o bidimensionales como el QR o cualquier otra que el fabricante considere y podrá acompañar físicamente al dispositivo médico o dirigir al usuario a donde se tenga acceso a la información sanitaria del etiquetado (a través de un sitio web o cualquier otra plataforma electrónica) sin que esta sea sustituida o contradictoria con lo autorizado.
5.3 Lo señalado en la presente Norma corresponde a la información sanitaria que debe ostentar el etiquetado de los dispositivos médicos, permitiéndose la inclusión de información adicional siempre que no se preste a confusión y que corresponda con las características del dispositivo médico y con la información relativa al registro sanitario otorgado por la COFEPRIS.
5.4 Los elementos del etiquetado de dispositivos médicos deben ser desarrollados y evaluados con base en una gestión de riesgos.
5.5 Proyectos de etiqueta para dispositivos médicos
La información contenida en las etiquetas o contraetiquetas debe corresponder a lo expresado en el oficio de registro sanitario autorizado, por la Secretaría de Salud, a través de la COFEPRIS, de conformidad con las disposiciones aplicables.
5.6 La información sanitaria contenida en los manuales e instrucciones de uso, son responsabilidad del fabricante y del titular del registro sanitario, debiendo cumplir con lo establecido en la presente Norma y las demás disposiciones aplicables.
5.7 Cuando la información sanitaria se exprese en otro idioma además del español, podrá ser hasta del mismo tamaño y proporcionalidad tipográfica, sin oponerse ni contravenir al texto en el idioma español.
5.7.1 Cuando la etiqueta de origen no contenga la información sanitaria establecida en la presente Norma, la información complementaria o faltante debe colocarse en la contraetiqueta de manera clara, legible, en un lugar visible y no debe cubrir la información sanitaria que comprometa la calidad del dispositivo médico, su uso o ambas.
5.7.1.1 Las contraetiquetas deben contener la información sanitaria complementaria establecida en la presente Norma y aquella determinada por la Secretaría de Salud, a través de la COFEPRIS. En el caso de dispositivos médicos de importación, esta podrá ser incorporada en territorio nacional, después del despacho aduanero, previo a su comercialización o suministro al público.
5.8 El medio, el formato, el contenido, la legibilidad, la integridad, la ubicación y el entorno del etiquetado deben ser apropiados de acuerdo con la naturaleza del dispositivo médico, en particular su indicación de uso y los usuarios previstos (el profesional de la salud y la población en general o ambas), para garantizar el uso seguro y eficaz.
5.8.1 En la información sanitaria del etiquetado se debe considerar el conocimiento técnico, la experiencia, la educación, la capacitación de los usuarios previstos, así como cualquier necesidad especial de las personas a quienes está destinado, la ubicación y el entorno en que se puede utilizar, además del uso coherente de la terminología.
5.8.2 El etiquetado debe estar sujeto al control de documentos (control de versiones) por parte del titular o fabricante.
5.9 Para el uso de símbolos en el etiquetado se deben utilizar los contenidos en el Apéndice A Normativo, y se podrá hacer uso de otro tipo de símbolos, siempre que la seguridad del dispositivo médico no se vea comprometida por la falta de comprensión por parte del usuario. Cuando el significado del símbolo no es obvio o no esté incluido en el Apéndice A Normativo de la presente Norma o en referencias internacionales, se debe proporcionar la descripción dentro del etiquetado y se debe evaluar mediante la gestión de riesgos.
5.10 Identificación del dispositivo médico
La identificación de los dispositivos médicos debe cumplir con lo dispuesto en los siguientes incisos, dependiendo de sus características y naturaleza. Se debe utilizar la leyenda o el símbolo alusivo del Apéndice A Normativo de la presente Norma.
5.10.1 Denominación genérica del dispositivo médico
El dispositivo médico debe ostentar una denominación genérica, que es el nombre que describe a un dispositivo médico o grupo de dispositivos médicos que tienen características comunes, aceptado por la COFEPRIS.
5.10.2 Denominación distintiva del dispositivo médico
El dispositivo médico debe identificarse mediante el uso de una denominación distintiva con la marca o el nombre comercial, que permita diferenciarlo de otros productos del mismo tipo o similar. Este es el único requisito que se permite se exprese en otro idioma diferente del español, si es el caso.
5.10.3 Número de registro sanitario otorgado por la COFEPRIS.
La etiqueta o contraetiqueta, debe indicar el número del registro sanitario otorgado por la COFEPRIS.
5.10.4 Fecha de caducidad del dispositivo médico
5.10.4.1 Se debe establecer en el etiquetado de los dispositivos médicos que por sus características e indicación de uso lo requieran. No debe exceder a la fecha de lo declarado por el fabricante de acuerdo con los estudios de estabilidad y esta fecha debe expresarse como año, mes; y cuando por la naturaleza de los dispositivos médicos lo requieran debe incluirse el día.
5.10.4.2 Para su identificación deben utilizarse las siguientes leyendas alusivas: "Caducidad___" o "Expiración___" o "Vencimiento___" o "Cad.___" o "Exp.___" o "Venc.___" u otra análoga, o el símbolo alusivo del Apéndice A Normativo de la presente Norma.
5.10.5 Fecha de fabricación
Debe incluir la fecha de fabricación; está puede incluirse como parte del lote o número de serie, siempre que la fecha sea claramente identificable.
5.10.6 Número de lote o número de serie
En cualquier parte del envase o empaque primario, secundario, múltiple o colectivo (siempre y cuando este último exista), debe figurar en todos los dispositivos médicos objeto de la presente Norma la identificación del número de lote o número de serie; debe aparecer con una indicación en clave o en lenguaje claro, ya sea grabado, marcado con tinta indeleble o de cualquier otro modo similar, establecido por el propio fabricante; para su identificación deben utilizarse las siguientes leyendas alusivas: "Lote ___" o "Número de serie ___" o "Lot. ___" o "Serie No. ___" u otra análoga o el símbolo alusivo del Apéndice A Normativo de la presente Norma.
5.10.7 Número de catálogo o control o referencia o modelo o código o número de versión del dispositivo médico
5.10.7.1 Un dispositivo médico, debe distinguirse de otros similares mediante el uso de un número de catálogo o código o control o referencia o modelo o número de versión, u otro método que permita la identificación del dispositivo médico con base en sus características distintivas, establecidas por el fabricante. Esta identificación debe guardar relación con una característica o especificación definida del dispositivo médico.
5.10.7.2 La distinción de un dispositivo médico de otros similares, podrá figurar en cualquier parte del envase o empaque primario o secundario o múltiple o colectivo, pudiendo utilizarse las siguientes leyendas: "Número de catálogo____" o "Control___" o "Referencia___", "Modelo___" o "Versión Número___", "No. Cat.___" o "Ctrl.___" o "REF.___", "Modelo___" o "Versión No.___" u otros análogos, o el símbolo alusivo del Apéndice A Normativo de la presente Norma.
5.10.8 Contenido del dispositivo médico
5.10.8.1 En el etiquetado del dispositivo médico debe indicar el número de unidades, piezas o elementos que lo componen, las dimensiones nominales, el contenido neto expresado en peso o volumen o incluido el volumen después de la reconstitución, número de pruebas o aplicaciones, según aplique en cada caso.
5.10.8.2 Para el caso de dispositivos médicos en envases o empaques múltiples o colectivos, éstos deben declarar el contenido en función de su denominación genérica.
5.10.8.3 En las instrucciones de uso se deben indicar aquellas limitaciones de uso en relación con el dispositivo médico, las precauciones relacionadas con los materiales incorporados al dispositivo médico, que sean potencialmente cancerígenos, mutágenos o tóxicos, o que puedan provocar sensibilización o reacción alérgica en el paciente o el usuario y se debe utilizar la leyenda o el símbolo alusivo del Apéndice A Normativo de la presente Norma.
5.10.9 Advertencias, precauciones o medidas
5.10.9.1 La etiqueta debe incluir todas las advertencias o precauciones que deban tomarse en cuenta y que deban comunicarse inmediatamente al usuario del dispositivo médico, y a cualquier otra persona cuando corresponda, con las siguientes leyendas de manera enunciativa, más no limitativa, "PRECAUCIÓN: SUPERFICIE CALIENTE" o "ESTE DISPOSITIVO MÉDICO CONTIENE LÁTEX" o "CONTIENE MATERIAL POTENCIALMENTE INFECCIOSO" o medidas que se deban adoptar y las limitaciones de uso. Esta información puede mantenerse al mínimo; se podrán utilizar los símbolos del apéndice A Normativo de la presente Norma, en cuyo caso deberá aparecer información más detallada en las instrucciones de uso o el manual.
5.10.9.2 Cuando el uso previsto de un dispositivo médico represente algún riesgo residual para algún grupo especialmente vulnerable de pacientes como puede ser el caso de niños o mujeres embarazadas o en período de lactancia, en las instrucciones de uso se debe incluir la información correspondiente y las medidas de precaución y advertencias necesarias.
5.10.9.3 Para el caso de dispositivos médicos que contengan sustancias tóxicas o peligrosas, deben llevar las leyendas de advertencia, informes de precaución o el símbolo alusivo del Apéndice A Normativo de la presente Norma, y deben sujetarse a lo establecido en la regulación vigente correspondiente.
5.10.9.4 Para el caso de los dispositivos médicos en los que intervengan fuentes de radiación se debe declarar en el etiquetado la leyenda: "PELIGRO, MATERIAL RADIACTIVO PARA USO EXCLUSIVO EN MEDICINA"; o el símbolo alusivo del Apéndice A Normativo de la presente Norma para la indicación de los isótopos que contienen actividad, vida media de los mismos y tipo de radiaciones que emiten.
5.10.9.5 Para los dispositivos médicos estériles se deben incluir, de manera enunciativa, más no limitativa, las leyendas siguientes u otras análogas, tales como:
· "DISPOSITIVO MÉDICO ESTÉRIL";
· "NO SE GARANTIZA LA ESTERILIDAD DEL DISPOSITIVO MÉDICO EN CASO DE QUE EL EMPAQUE PRIMARIO TENGA SEÑALES DE HABER SUFRIDO RUPTURA PREVIA".
O el símbolo alusivo del Apéndice A Normativo de la presente Norma.
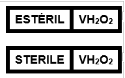
5.10.9.5.1 Se deben incluir las leyendas de manera enunciativa, más no limitativa, los procesos de esterilización u otras análogas, tales como:
· "ESTERILIZADO CON ÓXIDO DE ETILENO";
· "ESTERILIZADO CON RADIACIÓN GAMMA";
· "ESTERILIZADO CON CALOR SECO O HÚMEDO";
· "PROCESADOS USANDO TÉCNICAS ASÉPTICAS".
O el símbolo alusivo, con base en el Apéndice A Normativo de la presente Norma.
5.10.9.6 Cuando proceda, se deben colocar de manera enunciativa, más no limitativa, las siguientes leyendas alusivas a la seguridad del dispositivo médico: "ATÓXICO", "LIBRE DE PIRÓGENOS", o el símbolo alusivo del Apéndice A Normativo de la presente Norma.
5.10.9.7 Para los dispositivos médicos que son de un solo uso, se debe señalar dicha situación colocando las leyendas "DESECHABLE", "USAR SOLAMENTE UNA VEZ" o "DE UN SOLO USO", u otras leyendas o símbolos alusivos del Apéndice A Normativo de la presente Norma.
5.10.9.8 Si el dispositivo médico es reutilizable, en el etiquetado se debe informar sobre los procedimientos apropiados para su reutilización, incluida la limpieza, desinfección, descontaminación, embalaje, esterilización, cuando corresponda el método de reesterilización, así como el número de veces que sea posible su reutilización.
5.10.9.9 Se debe incluir en el etiquetado, la orientación sobre a quién debe contactar el paciente o usuario en caso de un incidente o incidente adverso (IA) con el dispositivo médico, incluyendo a la autoridad competente con base en lo establecido en el inciso 2.4 del Capítulo de Referencias Normativas de la presente Norma.
5.10.9.10 En el etiquetado se deben indicar las advertencias, precauciones o medidas que deben adoptarse frente a la exposición a influencias externas o condiciones ambientales razonablemente previsibles, como campos magnéticos, efectos eléctricos y electromagnéticos, descargas electrostáticas, radiaciones asociadas a procedimientos diagnósticos o terapéuticos, presión, humedad o temperatura, mediante el uso de leyendas o los símbolos alusivos del Apéndice A Normativo de la presente Norma.
5.10.9.11 En el etiquetado se debe incluir qué hacer en caso de que el envase se dañe o se abra accidentalmente antes de su uso, o si el envase se expone a condiciones ambientales diferentes de las especificadas.
5.10.9.12 El etiquetado no debe contener información con respecto a la responsabilidad del fabricante en caso de daños o lesiones resultantes de cualquier uso o mal funcionamiento del dispositivo médico, que contradiga la normatividad vigente.
5.10.9.13 El etiquetado no debe contener exenciones de responsabilidad relacionadas con la seguridad y desempeño del dispositivo médico para su uso previsto, que sean incompatibles con la normatividad vigente, o las obligaciones del fabricante de diseñar y fabricar un dispositivo médico que cumpla con los requerimientos de calidad, seguridad y desempeño, según lo previsto durante su vida útil esperada.
5.10.10 Indicación de uso del dispositivo médico
El fabricante o Titular del Registro Sanitario deben declarar la indicación de uso de los dispositivos médicos y las condiciones normales de uso en las cuales sean aptos, a través del etiquetado.
5.10.11 Instrucciones de uso
5.10.11.1 Las instrucciones de uso se deben proporcionar en términos comprensibles, tipografía y tamaño legibles para el usuario; puede complementarse de manera electrónica a través de RFID, o códigos de barras o bidimensionales como el QR o cualquier otra que el fabricante considere y podrá acompañar físicamente al dispositivo médico o dirigir al usuario a donde se tenga acceso a las instrucciones de uso (a través de un sitio web o cualquier otra plataforma electrónica) sin que esta sea sustituida o contradictoria con lo autorizado.
5.10.11.2 Las instrucciones de uso deberán incluir cualquier especificación que requiera el usuario para utilizar, procesar y mantener al dispositivo médico adecuadamente.
5.10.11.3 Las instrucciones de uso deben incluir los detalles sobre cualquier tratamiento preparatorio o de manipulación del dispositivo médico antes de que esté listo para su uso (de manera enunciativa, más no limitativa, esterilización, la identificación de otros equipos que no se proporcionan con el dispositivo médico, ensamble final, configuración, reconstitución o calibración).
5.10.11.4 Las instrucciones de uso deben incluir cualquier requisito relacionado con instalaciones especiales (de manera enunciativa, más no limitativa, campo estéril o cuartos limpios) o la capacitación especial o calificaciones particulares del usuario, o de terceros o ambos.
5.10.11.5 Las instrucciones de uso deben contener la información necesaria para comprobar si el dispositivo médico está correctamente instalado y listo para funcionar de manera segura y en la forma prevista por el fabricante, además de, cuando proceda:
· información y frecuencia del mantenimiento preventivo;
· información de la limpieza, la desinfección y disposición final, lo anterior sin perjuicio de otras obligaciones que, en su caso, impongan otras dependencias competentes;
· identificación de los componentes consumibles y cómo reemplazarlos;
· información de la calibración necesaria, y
· métodos para mitigar los riesgos encontrados durante la limpieza, la instalación, la calibración o el mantenimiento.
5.10.11.6 En caso de que el dispositivo médico o sus accesorios requieran de un manejo especial para su disposición final, las instrucciones de uso deben incluir toda advertencia o precaución que se debe adoptar en relación con ésta. Esto también incluye cualquier consumible que requiere un manejo especial como resultado de su uso con el dispositivo médico. Esta información debe abordar, cuando proceda:
· los peligros de infección o microbianos (de manera enunciativa, más no limitativa, explantes, agujas o equipo quirúrgico contaminados con sustancias de origen humano potencialmente infecciosas);
· los peligros ambientales (de manera enunciativa, más no limitativa; baterías o materiales que emiten niveles de radiación potencialmente peligrosos);
· los peligros físicos (de manera enunciativa, más no limitativa; objetos punzante).
Lo anterior sin perjuicio de otras obligaciones que, en su caso, impongan otras dependencias competentes.
5.10.11.7 Si el dispositivo médico se suministra sin esterilizar, con la intención de que se esterilice antes de su uso, se deben incluir las indicaciones para la esterilización, así como las instrucciones para limpiar, el dispositivo médico antes de la esterilización.
5.10.11.8 En el caso de los dispositivos médicos destinados a usarse junto con otros dispositivos médicos, las instrucciones de uso deben incluir información suficiente para identificar dichos dispositivos médicos, con el fin de obtener una combinación segura e información sobre cualquier restricción conocida a las combinaciones de éstos.
5.10.11.9 Si el dispositivo médico emite niveles peligrosos o potencialmente peligrosos de radiación para fines médicos, las instrucciones de uso deben incluir información detallada sobre la naturaleza, el tipo y, cuando proceda, la intensidad, la distribución y la dosis recomendada de la radiación emitida, y los medios para proteger al paciente, usuario o tercero de la radiación no intencional durante el uso del dispositivo médico, y las formas de evitar usos indebidos y de reducir los riesgos derivados de la instalación en la medida que sea posible y apropiado. Se debe utilizar la leyenda o el símbolo alusivo del Apéndice A Normativo de la presente Norma.
5.10.11.10 Las instrucciones de uso podrán indicar la fecha de emisión o última revisión y, en su caso, un número de identificación conforme al Sistema de Gestión de Calidad del fabricante o del Titular del Registro Sanitario.
5.10.11.11 Las instrucciones de uso deben contener el nombre y la dirección del fabricante o fabricante legal en un formato que sea reconocible y permita establecer la ubicación del fabricante o fabricante legal, junto con la información de contacto (de manera enunciativa, más no limitativa, número de teléfono o sitio web o dirección de correo electrónico), para obtener asistencia técnica.
5.10.12 Instrucciones de uso para agentes de diagnóstico in vitro
5.10.12.1 La descripción del uso previsto del agente de diagnóstico in vitro debe estar de conformidad con lo establecido en el inciso 2.6 del Capítulo de referencias normativas y, las instrucciones de uso deben incluir lo siguiente, cuando corresponda:
· tipo de prueba (de manera enunciativa, más no limitativa, prueba cualitativa, semicuantitativa, prueba cuantitativa);
· los parámetros que detecta o determina (de manera enunciativa, más no limitativa, glucosa, colesterol, triglicéridos, hepatitis, etc.);
· su función (de manera enunciativa, más no limitativa, cribado, seguimiento, diagnóstico o ayuda al diagnóstico, pronóstico, predicción, diagnóstico complementario);
· el trastorno, condición o factor de riesgo específico de interés que se pretende detectar, definir o diferenciar;
· si incluye o no componentes automatizados o si está destinado a ser utilizado con instrumentos automatizados;
· el tipo de muestra(s) (de manera enunciativa, más no limitativa, suero, plasma, sangre total, biopsia de tejido, orina) necesaria, incluida la fuente de la muestra (así como, sangre entera, capilar del brazo), matriz (de manera enunciativa, más no limitativa, tubo con EDTA), tiempo (a modo de ejemplo, 8 horas después de la lesión) y método de recolección (tal como, orina recolectada por uno mismo); y
· población objetivo en la que se utiliza.
5.10.12.2 Las instrucciones de uso deben incluir todos los procedimientos de control de calidad necesarios para comprobar que el agente de diagnóstico in vitro funciona en la forma prevista, incluyendo los siguientes, según corresponda:
· los procedimientos para utilizar los controles disponibles;
· las instrucciones que recomienden la frecuencia de uso;
· las limitaciones del procedimiento de control de calidad;
· una explicación de la manera en la que el usuario debe interpretar los resultados del procedimiento de control de calidad, incluyendo una descripción que le permita decidir cuándo SÍ o cuando NO pueden aceptarse los resultados de la prueba; y
· las medidas que deben adoptarse en caso de falla de alguno de los controles.
5.10.12.3 Las instrucciones de uso deben incluir una declaración de los principios de la prueba, de manera enunciativa, más no limitativa, aspecto biológico, químico, microbiológico, inmunoquímico y otros en los que se basen. La información que se considere confidencial no requiere ser divulgada, pero deben proporcionarse suficientes detalles para que el usuario comprenda cómo el dispositivo médico puede realizar su función.
5.10.12.4 Las instrucciones de uso deben incluir una descripción y la cantidad de reactivos, calibradores, controles y toda limitación sobre su uso (de manera enunciativa, más no limitativa, adecuado solamente para un instrumento).
5.10.12.5 Los kits de agentes de diagnóstico in vitro, que incluyen reactivos individuales y materiales que se encuentren disponibles como dispositivos médicos separados deben de cumplir con las instrucciones de uso de conformidad con esta sección.
5.10.12.6 Las instrucciones de uso deben incluir una lista de los materiales proporcionados y de cualquier material requerido, pero no proporcionado.
5.10.12.7 Las instrucciones de uso deben incluir una descripción de la estabilidad en uso. Esta descripción puede incluir las condiciones de almacenamiento antes de abrir el envase o empaque primario, junto con las condiciones de almacenamiento y estabilidad de las soluciones de trabajo, cuando proceda.
5.10.12.8 Las instrucciones de uso deben contener las condiciones incluidas y excluidas para la recolección, el envío, el manejo y la preparación de la muestra.
5.10.12.9 En las instrucciones de uso o en alguno de los elementos del etiquetado deben indicar la trazabilidad de valores asignados a calibradores y a los materiales empleados para la verificación de la calibración, incluyendo identificación de materiales de referencia aplicable o procedimientos de medición de referencia de orden superior.
5.10.12.10 Las instrucciones de uso deben describir el procedimiento de la prueba, incluidos los cálculos e interpretación de los resultados, todo software o base de datos de referencia adicionales que sean necesarios y, en su caso, si es necesario considerar la posibilidad de una prueba de confirmación.
5.10.12.11 En las instrucciones de uso o en alguno de los elementos del etiquetado deben listar las características del desempeño analítico, de manera enunciativa, más no limitativa, precisión, exactitud, sensibilidad y especificidad.
5.10.12.12 En las instrucciones de uso o en alguno de los elementos del etiquetado de acuerdo con la naturaleza del dispositivo médico y uso previsto deben enumerar las características del desempeño clínico (de manera enunciativa, más no limitativa, sensibilidad diagnóstica, especificidad diagnóstica, valor predictivo positivo, valor predictivo negativo, razón de probabilidad, valores esperados en poblaciones normales y afectadas).
5.10.12.13 Las instrucciones de uso deben incluir información sobre cualquier sustancia interferente o limitaciones (de manera enunciativa, más no limitativa, evidencia visual de hiperlipidemia o hemólisis, edad de la muestra) que puedan alterar el desempeño del análisis.
5.10.12.14 Cuando proceda, las instrucciones de uso deben incluir una sección de bibliografía o referencias.
5.10.13 Riesgos residuales
5.10.13.1 Los riesgos residuales deben incluirse con base en la gestión de riesgos, en las instrucciones de uso o en cualquier elemento del etiquetado y se consideran información de seguridad.
5.10.13.2 Se puede incluir en las instrucciones de uso o colocar una referencia donde se tenga acceso al resumen de los estudios de desempeño y las investigaciones clínicas utilizadas para demostrar la conformidad con la normatividad aplicable y que demuestren la seguridad y el desempeño clínico del dispositivo médico o de un agente de diagnóstico in vitro para su uso previsto, este puede incluir, entre otros, un resumen de la investigación, el desempeño clínico y los datos de resultados, información de seguridad clínica y un resumen del beneficio clínico del dispositivo médico o de un agente de diagnóstico in vitro, y puede presentarse de tal manera que refleje con precisión la seguridad y el desempeño del dispositivo médico o de un agente de diagnóstico in vitro.
5.10.13.3 Se debe incluir en las instrucciones de uso la información sobre los materiales, sustancias y los residuos de fabricación que representen un riesgo para la salud del paciente. Se debe utilizar la leyenda o el símbolo alusivo del Apéndice A Normativo de la presente Norma.
5.10.14 Manejo y almacenamiento del dispositivo médico
Las instrucciones de uso deben incluir toda medida especial de manipulación o condiciones ambientales aceptables (tales como, los límites superiores e inferiores de temperatura, luz, humedad) para el almacenamiento y el transporte del dispositivo médico. Debe evitarse el uso de indicaciones de temperatura o humedad no específicas que estén abiertas a interpretación, o que puedan variar según la ubicación geográfica, a menos que se agreguen condiciones complementarias. Se debe utilizar la leyenda o el símbolo alusivo del Apéndice A Normativo de la presente Norma.
5.11 Identificación del fabricante del dispositivo médico
Las modalidades para la expresión de las condiciones de fabricación y comercialización deben ser las siguientes o sus análogas. Se debe utilizar la leyenda o el símbolo alusivo del Apéndice A Normativo de la presente Norma.
5.11.1 Cuando el fabricante del dispositivo médico en México sea nacional se expresará(n) la(s) leyenda(s) que aplique(n):
5.11.1.1 "Fabricado en México por:" seguido de la razón social y domicilio.
5.11.1.2 "Acondicionado por:" seguido de la razón social y domicilio del establecimiento.
5.11.1.3 "Fabricado para:" seguido de la razón social y domicilio.
5.11.1.4 "Distribuido por:" seguido de la razón social y domicilio del establecimiento.
5.11.2 Para los dispositivos médicos importados se expresarán la(s) leyenda(s) que aplique:
5.11.2.1 "Fabricado por:" seguido de la razón social y domicilio.
5.11.2.2 "Fabricado para:" seguido de la razón social y domicilio.
5.11.2.3 "Importado por:" seguido de la razón social y domicilio del establecimiento.
5.11.2.4 "Acondicionado por:" seguido de la razón social y domicilio del establecimiento.
5.11.2.5 "Distribuido por:" seguido de la razón social y domicilio del establecimiento. Para el caso en que la razón social y domicilio del importador coincida con el distribuidor, se podrá incluir el texto "Importado y Distribuido por:" seguido de la razón social y del domicilio del establecimiento.
5.11.2.6 Colocar la leyenda alusiva que identifique el país de origen del dispositivo médico o gentilicio de acuerdo con lo dispuesto en la normatividad vigente y los tratados internacionales de los cuales México forma parte. No debe confundirse el término "fabricante" aprobado en el registro sanitario con el de "país de origen", éste puede ser distinto.
5.11.3 El domicilio debe contener los siguientes datos o su equivalente cuando aplique: calle/carretera, número/casa/piso, colonia, ciudad, estado, código postal y país. Podrán utilizarse abreviaturas.
5.12 El etiquetado de los dispositivos médicos que estén destinados a muestras sin fines de lucro deben estar identificadas conforme al SGC e incluir la leyenda "prohibida su venta".
5.13 Desempeño del dispositivo médico
5.13.1 Se deben incluir en las instrucciones de uso las precauciones u otras medidas que debe tomar el paciente si el desempeño del dispositivo médico cambia o el paciente experimenta cualquiera de los signos de falla del mismo.
5.13.2 Con base en la naturaleza del dispositivo médico o su indicación de uso en el etiquetado se debe indicar, la vida útil esperada del dispositivo médico y cualquier factor que pueda afectarlo.
5.13.3 Se deben incluir en las instrucciones de uso las precauciones u otras medidas que deben tomarse al final de la vida útil esperada o cerca del mismo.
6. Requisitos específicos para el etiquetado de los dispositivos médicos
6.1 Para el etiquetado de este tipo de dispositivos médicos adicional a lo indicado en el Capítulo 5 de la presente Norma, se debe considerar lo siguiente:
6.1.1 La información sanitaria específica debe considerar lo descrito en los apartados señalados como "etiquetado" dentro de la monografía del producto, de conformidad con lo establecido en el inciso 2.6 del Capítulo de Referencias Normativas de la presente Norma.
6.1.2 En alguno de los elementos del etiquetado de los dispositivos médicos destinados exclusivamente para las instituciones públicas de salud y de seguridad social, deben incluir al menos una de las leyendas "Prohibida su venta" o "Propiedad del Sector Salud".
6.1.3 Para radiofármacos se debe indicar la fecha y hora de caducidad, que marcan el fin de su vida útil con base en los estudios de estabilidad.
6.1.4 Para dispositivos médicos considerados como juego/paquete (kit)
6.1.4.1 Deben tener en su etiqueta o contraetiqueta el número de registro sanitario del juego/paquete (kit), declarar los componentes que integran dicha presentación y cuando aplique el número de Registro Sanitario de cada insumo para la salud.
6.1.4.2 Cuando el juego/paquete (kit), contenga dispositivos médicos que también se comercialicen de forma individual cada uno de ellos deberá cumplir con los requisitos del etiquetado establecidos en la presente Norma.
6.1.4.3 En la etiqueta del juego/paquete (kit) se debe indicar la fecha de caducidad del dispositivo médico que ostente el menor periodo de caducidad.
6.1.4.4 Se debe colocar un número de lote diferente y específico para el juego/paquete (kit) en el empaque en el que se suministre.
6.1.5 Para dispositivos médicos formulados
6.1.5.1 Se debe declarar en el etiquetado la fórmula cualitativa o sus principios activos o fármacos contenidos.
6.1.6 Para los dispositivos médicos que contengan o lleven incorporado: Células o tejidos, o sus derivados, de origen humano; Células o tejidos, o sus derivados de origen animal; Derivados de sangre o plasma humanos, o fármaco o medicamento.
6.1.6.1 Se debe indicar en el etiquetado si el dispositivo médico contiene o incorpora un fármaco, medicamento o una sustancia biológica, incluir la cantidad, proporción o concentración de la misma y si estará en contacto directo con el paciente.
6.1.7 En el etiquetado de los medios de cultivo en polvo, debe incluirse el método de preparación y el pH final.
6.1.8 Para Agentes de diagnóstico in vitro
6.1.8.1 Se debe incluir en el etiquetado información sanitaria que describa en qué momento no debe utilizarse más el agente de diagnóstico in vitro, como en el caso de signos de degradación o el número máximo de reutilizaciones. Se debe utilizar la leyenda o el símbolo alusivo del Apéndice A Normativo de la presente Norma.
6.1.8.2 Se deben indicar en el etiquetado las advertencias, precauciones o medidas que deben adoptarse en caso de mal funcionamiento o degradación del agente de diagnóstico in vitro, indicadas por cambios de aspecto que puedan afectar al funcionamiento.
6.1.8.3 Las etiquetas y contraetiquetas de los agentes de diagnóstico in vitro que se empleen en dispositivos médicos deben incluir la leyenda "agente de diagnóstico in vitro" y el símbolo correspondiente a la leyenda, del Apéndice A Normativo de la presente Norma, y "para uso exclusivo en laboratorios clínicos o de gabinetes" sólo cuando se emplean en dispositivos médicos ubicados en laboratorios clínicos o unidades de laboratorio en hospitales.
6.2 Para dispositivos médicos con limitaciones de tamaño o diseño
En aquellos dispositivos médicos que, debido a su naturaleza, tamaño o diseño, la etiqueta no pueda contener la información especificada en el Capítulo 5 de la presente Norma, estos dispositivos médicos deben contener al menos número de lote, denominación genérica, denominación distintiva, contenido, excepto cuando éste sea obvio y la fecha de caducidad en el envase o empaque primario. La información sanitaria complementaria o faltante que se señala en el Capítulo 5 de la presente Norma, debe aparecer en alguno de los elementos del etiquetado.
6.3 Software como dispositivo médico (ScDM)
6.3.1 El software como dispositivo médico (ScMD) debe identificarse con un identificador, tal como la versión, nivel de revisión o fecha de lanzamiento/edición. El identificador único debe ser accesible para el usuario previsto, a menos que el dispositivo médico no tenga una interfaz electrónica alámbrica o inalámbrica.
6.3.2 Para software sin forma física o empaque, el etiquetado puede estar disponible en forma electrónica. En esta situación, debe incorporar un medio para que el usuario acceda fácilmente al etiquetado electrónico a través del propio software o mediante la inclusión de un sitio web u otros medios.
6.4 Requisitos específicos de etiquetado para dispositivos médicos para uso de la población en general
6.4.1 La información sanitaria y las instrucciones de uso contenidas en el etiquetado proporcionadas por el fabricante deben estar en términos comprensibles para que la población en general las aplique correctamente.
6.4.2 Las instrucciones de uso destinadas principalmente a población en general deben redactarse en términos fácilmente comprensibles para el usuario previsto y, en su caso, complementarse con dibujos y diagramas junto al texto correspondiente y estar disponibles en un formato apropiado y accesible para el usuario.
6.4.3 Las instrucciones facilitadas por el fabricante deben permitir a la población en general comprenderlas y aplicarlas, a fin de interpretar correctamente el resultado proporcionado por el dispositivo médico o confirmar que el dispositivo médico funciona o ha funcionado según el uso previsto.
6.4.4 En las instrucciones de uso se puede simplificar la indicación de uso, siempre que se mantenga la información fundamental, pueden omitir algunos de los elementos recomendados, siempre que esto no altere la seguridad o el desempeño y la justificación de cualquier omisión debe describirse en la gestión de riesgos por el fabricante del dispositivo médico.
6.4.5 Las instrucciones de uso deben describir de manera clara y concisa las circunstancias en las que el usuario debe consultar con un profesional de la salud.
6.4.6 Las instrucciones de uso deben indicar claramente si un agente de diagnóstico in vitro está destinado para autopruebas y si requiere la participación de un tercero que brinde cuidados para su aplicación.
6.4.7 El fabricante debe informar cualquier contra indicación, advertencia o precaución que deba tomarse para apoyar y asistir a los usuarios del dispositivo médico en su uso seguro y apropiado.
7. Concordancia con Normas Internacionales
La presente Norma Oficial Mexicana no es equivalente (NEQ) con alguna Norma Internacional.
8. Bibliografía
8.1 Guidance for industry and FDA staff. Use of symbols on labels and in labeling of in vitro diagnostic devices intended for professional use.- Washington, Food and Drug Administration, November, 2004.
8.2 Code of Federal Register Part 91-4179 Medical Device Good Manufacturing Practices Manual.- Washington, Food and Drug Administration, April, 2004.
8.3 Global Harmonization Task Force. GHTF/SG1/N43:2005 Labelling for Medical Devices.- June 3, 2005.
8.4 Global Harmonization Task Force GHTF/SG1/N77:2012 Principles of Medical Devices Classification.- November 2nd, 2012.
8.5 Reglamento (UE) 2017/745 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 5 de abril de 2017 sobre los productos sanitarios, por el que se modifican la Directiva 2001/83/CE, el Reglamento (CE) no.178/2002 y el Reglamento (CE) no.1223/2009 y por el que se derogan las Directivas 90/385/CEE 93/42/CEE del CONSEJO, 02017R0745-ES-09.07.2024.
8.6 Reglamento (UE) 2017/746 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 5 de abril de 2017 sobre los productos sanitarios para diagnóstico in vitro y por el que se derogan la Directiva 98/79/CE y la Decisión 2010/227/UE de la Comisión, 02017R0746-ES-09.07.2024.
8.7 ISO 15223-1:2021 Medical devices- Symbols to be used with information to be supplied by the manufacturer- Part 1: General requirements, Fourth edition, 07, 2021.
8.8 ISO 15223-2:2010 Medical devices-Symbols to be used with medical device labels, labelling, and information to be supplied-Part2: Symbols development, selection and validation, First edition, 01, 2010.
8.9 ISO 20417:2021 Medical Devices-Information to be supplied by the manufacturer, First edition, 2021-04.
8.10 ISO 7010:2019 Graphical symbols-Safety colours and safety signs-Registered safety signs, Third edition, 2019-07.
8.11 ISO 3864-1:2011 Graphical symbols - safety colours and safety signs -Part 1: Design principles for safety signs and safety markings, Second edition, 2011-04-15.
8.12 WHO Expert Committee on Biological Standardization: seventy-eighth report. Geneva: World Health Organization; 2024 (WHO Technical Report Series, No. 1054). Licence: CC BYNC-SA 3.0 IGO, ISBN 978-92-4-009183-2.
8.13 OPS/HSS/MT/22-0007 Principios de etiquetado de los dispositivos médicos y los dispositivos médicos de diagnóstico in vitro, 2022.
8.14 IMDRF/GRRP WG/N47 FINAL:2024 (Edition 2) Essential Principles of safety and Performance of Medical Devices and IVD Medical Devices, 26 April 2024.
8.15 IMDRF/GRRP WG/N52 FINAL:2024 (Edition 2) Principles of Labeling for Medical Devices and IVD Medical Devices, 26 April 2024.
8.16 IMDRF/AIMD WG/N67 Machine Learning-enabled Medical Devices: Key Terms and Definitions, 6 May 2022.
8.17 MD-MDCG 2021-26 Question and answers on repackaging & relabelling activities under Article 16 of Regulation (EU) 2017/745 and Regulation (EU) 2017/746, October 2021.
8.18 ISO/IEC GUIDE 63:2019(E), Guide to the development and inclusion of aspects of safety in International Standars for medical devices, Third edition, 2019-08.
8.19 ISO/IEC GUIDE 51:2014(E), Safety aspects -Guidelines for their inclusion in standards, Third edition, 2014-04-01.
8.20 ISO 14971:2019 Dispositivos médicos, aplicación de la Gestión de riesgos a los dispositivos médicos, edition 3, 2019-12.
8.21 ISO 3166-1:2020 Codes for the representation of names of countries and their subdivisions, Part 1: Country Code. edition 4, 2020-08.
8.22 21 CFR PART 801. (https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-801).
8.23 WHO/BS/2022.2425, WHO Global model regulatory framework for medical devices including in vitro diagnostic medical devices (GMRF), 2023.
8.24 Global Harmonization Task Force. GHTF/SG2/N54R8:2006, Medical Devices Post Market Surveillance: Global Guidance for Adverse Event Reporting for Medical Devices.- 30 November, 2006.
8.25 Orientaciones para la vigilancia poscomercialización y la vigilancia del mercado de los dispositivos médicos, incluidos los de diagnóstico in vitro [Guidance for post-market surveillance and market surveillance of medical devices, including in vitro diagnostics]; Ginebra: Organización Mundial de la Salud; 2021. Licencia: CC BY-NC-SA 3.0 IGO.
9. Observancia de la Norma
La vigilancia del cumplimiento de la presente Norma corresponde a la Secretaría de Salud, a través de la COFEPRIS, cuyo personal realizará la verificación y la vigilancia que sean necesarias.
10. Evaluación de la conformidad
10.1 La evaluación de la conformidad de la presente Norma lo realizará la Secretaría de Salud, a través de la Comisión Federal para la Protección contra Riesgos Sanitarios.
10.2 El procedimiento para la Evaluación de la Conformidad y a fin de determinar el grado de cumplimiento de la presente Norma se efectuarán verificaciones por parte de personal de la COFEPRIS, en cualquiera de las siguientes opciones:
10.2.1 En los sitios de fabricación de dispositivos médicos, a través del etiquetado o contraetiquetado en sus almacenes y en los del distribuidor, conforme a lo autorizado en el Oficio de Registro Sanitario y los puntos cubiertos en la presente Norma.
10.2.2 Durante la evaluación de la solicitud del Registro Sanitario de un dispositivo médico o sus modificaciones del mismo a través de las leyendas sanitarias que se presenten.
11. Vigencia
La presente Norma entrará en vigor a los 360 días naturales posteriores a su publicación en el Diario Oficial de la Federación.
TRANSITORIOS
PRIMERO.- La entrada en vigor de la presente Norma, deja sin efectos a la Norma Oficial Mexicana NOM-137-SSA1-2008, Etiquetado de dispositivos médicos, publicada en el Diario Oficial de la Federación el 12 de diciembre de 2008.
SEGUNDO.- Para los dispositivos médicos cuyo etiquetado no pueda modificarse la información sanitaria requerida en la Norma Oficial Mexicana NOM-137-SSA1-2025, Etiquetado de dispositivos médicos, tendrán 180 días naturales posteriores a la entrada en vigor de la presente Norma, para agotar la existencia de materiales de envases y productos terminados.
Ciudad de México a 11 de marzo de 2026.- El Comisionado Federal para la Protección contra Riesgos Sanitarios y Presidente del Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario, Victor Hugo Borja Aburto.- Rúbrica.
Apéndice A
(Normativo)
Símbolos para el etiquetado de los dispositivos médicos
Para la aplicación y compilación de los símbolos se recomienda considerar:
A.1 Cuando el fabricante, fabricante legal, importador, distribuidor y en su caso el acondicionador corresponde a la misma entidad responsable, solo será necesario describir el nombre y la dirección, una sola vez.
A.2 Criterios de uso de los símbolos
A.2.1 Cuando se utilicen los símbolos en el etiquetado de los dispositivos médicos, se recomienda considerar lo siguiente:
- Símbolo
- Título del símbolo
- Condiciones de uso del símbolo y la identidad de la audiencia propuesta
- Información sobre cualquier símbolo relacionado existente o propuesto
A.2.2 Para maximizar el espacio real de la etiqueta para el uso de los símbolos, se sugiere desarrollar una agrupación u orden consistente para el etiquetado, la manifestación de la marca, planificación de futuros idiomas que se agregarán y agruparán los idiomas de manera que se ajuste al modelo de distribución de la empresa.
A.2.3 Con la intención de que el nombre y el domicilio de las figuras previamente mencionadas se asocien claramente a los símbolos, pueden utilizarse elementos gráficos adicionales. Si son de dos a cinco símbolos, estos pueden ser agrupados vertical u horizontalmente o aparecer agrupados de cualquier manera que no genere confusión.
A.2.4 El símbolo de prohibición general tiene por objeto indicar una acción prohibida. Para el etiquetado de dispositivos médicos, el círculo de prohibición con una barra diagonal debería utilizarse para indicar "no", tal como, el símbolo 38 "No contiene látex".
A.2.5 Es parte de la gestión de riesgos determinar el tamaño apropiado para que el símbolo sea legible para el usuario para la función prevista.
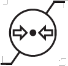
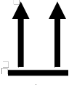
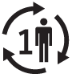
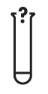
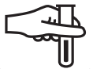
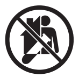
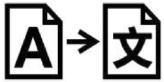
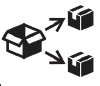
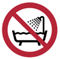
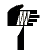
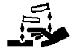
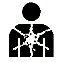
A continuación, se presenta la Tabla A.1 de los símbolos para el etiquetado de dispositivos médicos.
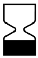
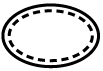
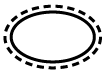
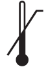
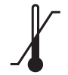
Tabla A.1 - Símbolos para el etiquetado de los dispositivos médicos
| NO.
| SÍMBOLO
| TÍTULO
| DEFINICIÓN |
| FABRICANTE
|
| 1
| | Fabricante
| Esté símbolo puede ir acompañado del nombre y dirección del fabricante, adyacente al símbolo. Se puede combinar el símbolo con la fecha de fabricación. |
| 2
| | Fecha de fabricación
| Indica la fecha en que se fabricó el dispositivo médico conforme al inciso 5.10.5, la fecha estará adyacente al símbolo. |
| 3
| | País del Fabricante
| Indica el país del fabricante del dispositivo médico. El "CC" debe sustituirse por el código del país de dos letras o el de tres letras conforme a la ISO 3166-1. |
| 4
| | Fecha de caducidad
| Indica la fecha en la que el dispositivo médico no debe ser usado conforme al inciso 5.10.4. La fecha se colocará adyacente al símbolo. |
| 5
| | Número de lote
| Indica el número de lote del fabricante conforme al inciso 5.10.6. El número de lote puede estar colocado adyacente al símbolo. |
| 6
| | Número de serie
| Indica el número de serie del fabricante conforme al inciso 5.10.6 de forma que identifica el dispositivo médico. El número de serie estará colocado adyacente al símbolo. |
| 7
| | Número de referencia
| Indica el número de referencia del fabricante conforme al inciso 5.10.7 de forma que identifica el dispositivo médico. El número de referencia estará adyacente al símbolo. |
| 8
| | Número de modelo
| Indica el número de modelo del fabricante conforme al inciso 5.10.7 de forma que identifica el dispositivo médico. El número de modelo estará adyacente al símbolo. |
| 9
| | Importador
| Indica el establecimiento que importa el dispositivo médico a la región. Este símbolo estará acompañado por el nombre y la dirección del establecimiento importador adyacente al símbolo. |
| 10
| | Distribuidor
| Indica el establecimiento que distribuye el dispositivo médico en la región. Este símbolo estará acompañado por el nombre y la dirección del establecimiento distribuidor adyacente al símbolo. |
| ESTERILIDAD
|
| 11
| | Estéril
| Indica un dispositivo médico que ha sido sometido a un proceso de esterilización. El uso de este símbolo excluye el uso de los símbolos 12 al 16. |
| 12
| | Esterilizado mediante técnicas
de procesamiento aséptico
| Indica un dispositivo médico que ha sido fabricado usando técnicas asépticas aceptadas. El uso de este símbolo excluye el uso del símbolo 11. |
| 13
| | Esterilizado con óxido de
etileno
| Indica un dispositivo médico que ha sido esterilizado con óxido de etileno. El uso de este símbolo excluye el uso del símbolo 11. |
| 14
| | Esterilizado mediante
irradiación
| Indica un dispositivo médico que ha sido esterilizado mediante irradiación. El uso de este símbolo excluye el uso del símbolo 11. |
| 15
| | Esterilizado con vapor o calor
seco
| Indica un dispositivo médico que ha sido esterilizado con vapor o calor seco. El uso de este símbolo excluye el uso del símbolo 11. |
| 16
| | Esterilizado con peróxido de
hidrógeno vaporizado
| Indica un dispositivo médico que se ha esterilizado con peróxido de hidrógeno vaporizado. El uso de este símbolo excluye el uso del símbolo 11. |
| 17
| | Vía de fluido estéril
| Indica la presencia de una vía de fluidos estériles dentro de un dispositivo médico en los casos en que otras partes del dispositivo médico, incluido el exterior, podrían no proporcionarse estériles. El método de esterilización se indicará en la casilla vacía del símbolo, según corresponda. La parte del dispositivo médico que es estéril se identificará en la información proporcionada por el fabricante. |
| 18
| | No reesterilizar
| Este símbolo solo se utilizará cuando este acompañado del símbolo estéril (11 al 16). Este símbolo no se utilizará en dispositivos médicos reutilizables que están indicados para esterilizarse entre usos. |
| 19
| | No estéril
| Indica un dispositivo médico que no ha sido sometido a un proceso de esterilización. Este símbolo solo debe usarse para distinguir entre dispositivos médicos idénticos o similares vendidos en condiciones estériles y no estériles. |
| 20
| | No usar si la barrera estéril
está dañada o si el empaque
está comprometido
| Indica que un dispositivo médico no se debería utilizar si su envase ha sido dañado o se ha abierto. |
| 21
| | Sistema de barrera estéril
único
| Indica un sistema de barrera estéril único. Este símbolo se colocará al lado o en combinación con el símbolo 11, 12, 13, 14, 15, 16 o 17. NOTA: una línea continua identifica un sistema de barrera estéril. |
| 22
| | Sistema de barrera estéril
doble
| Indica dos sistemas de barrera estéril. Este símbolo se colocará junto a o en combinación con el símbolo 11, 12, 13, 14, 15, 16 o 17. NOTA: una línea continua doble indica un sistema de barrera estéril doble. |
| 23
| | Sistema de barrera estéril
simple con embalaje protector
en el interior
| Indica un único sistema de barrera estéril con empaque protector en el interior. Este símbolo se colocará al lado o en combinación con el símbolo 11, 12, 13, 14, 15, 16 o 17. NOTA: el empaque protector situado dentro del sistema de barrera estéril está diseñado para evitar daños en el sistema de barrera estéril al contenido. La protección puede ser contra peligros físicos, contaminación de partículas u otros peligros ambientales, pero no incluye una barrera microbiana. |
| 24
| | Sistema de barrera estéril
simple con embalaje protector
en el exterior
| Indica un único sistema de barrera estéril con envase protector en el exterior. Este símbolo se colocará al lado o en combinación con el símbolo 11, 12, 13, 14, 15, 16 o 17. NOTA: el embalaje protector ubicado fuera del sistema de barrera estéril está diseñado para evitar daños al sistema de barrera estéril y al contenido. La protección puede ser contra peligros físicos, contaminación por partículas u otros peligros ambientales, pero no incluye una barrera microbiana. |
| ALMACENAMIENTO |
| 25
| | Frágil, manipular con cuidado
| Indica un dispositivo médico que puede romperse o dañarse si no se manipula con cuidado. |
| 26
| | Mantener alejado de la luz
solar o el calor
| Indica un dispositivo médico que necesita protección de la radiación solar y también "Mantener alejado de la luz solar o el calor". |
| 27
| | Proteger del calor y fuentes
radiactivas
| Indica un dispositivo médico que necesita protección de la luz solar y de fuentes radiactivas. NOTA: este símbolo también significa "Mantener alejado de la luz solar y de fuentes radiactivas". |
| 28
| | Mantener seco o alejado de la
lluvia
| Indica un dispositivo médico que debe protegerse de la humedad. NOTA: este símbolo también puede significar "Mantener alejado de la lluvia". |
| 29
| | Límite inferior de temperatura
| Indica el límite inferior de temperatura al que se puede exponer con seguridad el dispositivo médico. El límite inferior de temperatura se indicará junto a la línea horizontal inferior. |
| 30
| | Límite superior de temperatura
| Indica el límite superior de temperatura al que se puede exponer con seguridad el dispositivo médico. El límite superior de temperatura se indicará junto a la línea horizontal superior. |
| 31
| | Límite de temperatura
| Indica los límites de temperatura a los que se puede exponer el dispositivo médico de forma segura. Los límites de temperatura superior e inferior, se indicarán junto a las líneas horizontales superior e inferior. |
| 32
| | Limitación de humedad
| Indica el rango de humedad al que se puede exponer con seguridad el dispositivo médico. La limitación de humedad se indicará junto a las líneas horizontales superior e inferior. |
| 33
| | Limitación de la presión
atmosférica
| Indica el rango de presión atmosférica al que se puede exponer con seguridad el dispositivo médico. Las limitaciones de presión atmosférica se indicarán junto a las líneas horizontales superior e inferior. |
| USO SEGURO
|
| 34
| | No reutilizar
| Indica un dispositivo médico destinado a un solo uso. NOTA: los sinónimos de "No reutilizar" son "Un solo uso" y "Usar una sola vez". |
| 35
| | Consultar instrucciones de uso
| Indica la necesidad de que el usuario consulte las instrucciones de uso. |
| 36
| | Precaución
| Para indicar que es necesario tener precaución cuando se opera el dispositivo médico o el control cerca de donde se coloca el símbolo, o para indicar que la situación actual requiere que el operador sea consiente o actué para evitar consecuencias indeseables. |
| 37
| | Este lado hacia arriba
| Esta es la correcta posición hacia arriba de la distribución de empaques para el transporte, almacenamiento o ambos. |
| 38
| | Contiene o tiene presencia de
látex
| Indica la presencia de caucho natural seco o látex como material de construcción dentro del dispositivo médico o el envase de un dispositivo médico. NOTA: este símbolo pretende advertir a aquellas personas que pueden tener reacciones alérgicas a ciertas proteínas del látex. Este símbolo no debe usarse para dispositivos médicos que contengan "caucho sintético". |
| 39
| | Contiene sangre humana o
derivados del plasma
| Indica un dispositivo médico que contiene o incorpora sangre humana o derivados del plasma. |
| 40
| | Contiene un fármaco o
medicamento.
| Indica un dispositivo médico que contiene o incorpora un fármaco o medicamento. |
| 41
| | Contiene material biológico de
origen animal
| Indica un dispositivo médico que contiene tejido biológico, células o sus derivados, de origen animal (bovinos, ovinos, caprinos, ciervos, alces, visones, gatos). |
| 42
| | Contiene material biológico de
origen humano
| Indica un dispositivo médico que contiene tejido biológico, células o sus derivados, de origen humano. |
| 43
| | Contiene sustancias
peligrosas
| Indica un dispositivo médico que contiene sustancias que pueden ser cancerígenas, mutagénicas, reprotóxicas (CMR) o sustancias con propiedades de alteración endocrina. NOTA: el término "sustancias" se utiliza para indicar una sola sustancia o múltiples sustancias. |
| 44
| | Contiene nanomateriales
| Indica un dispositivo médico que contiene nanomateriales. |
| 45
| | Un solo Paciente - uso
múltiple
| Indica un dispositivo médico que se puede usar varias veces (procedimientos múltiples) en un solo paciente. |
| AGENTE DE DIAGNÓSTICO IN VITRO-ESPECIFICO
|
| 46
| | Agente de diagnóstico in vitro
| Indica un dispositivo médico que está destinado a ser utilizado como agente de diagnóstico in vitro. Este símbolo solo debe usarse para identificar los agentes de diagnóstico in vitro y no para especificar que el dispositivo médico es para "uso in vitro". |
| 47
| | Control
| Indica un material de control destinado a verificar el desempeño de otro dispositivo médico. NOTA: para controles negativos, utilice símbolo 48 y para controles positivos utilice el símbolo 49. |
| 48
| | Control negativo
| Indica un material de control destinado a verificar los resultados en el rango negativo esperado. |
| 49
| | Control positivo
| Indica un material de control destinado a verificar los resultados en el rango positivo esperado. |
| 50
| | Contiene suficiente para <n>
pruebas
| Indica el número total de pruebas in vitro que se puede realizar con el dispositivo médico. Junto al símbolo aparecerá el número de pruebas que se pueden realizar con el dispositivo médico. |
| 51
| | Solo para evaluación del
desempeño del agente de
diagnóstico in vitro
| Indica el agente de diagnóstico in vitro que está destinado a ser utilizado solo para evaluar el desempeño antes de comercializarlo para uso de diagnóstico médico. NOTA: un dispositivo médico destinado a ser utilizado únicamente para evaluar el desempeño del agente de diagnóstico in vitro antes de su comercialización para uso en diagnóstico médico Este símbolo no aparecerá conjuntamente con el símbolo 46. |
| TRANSFUSIÓN/INFUSIÓN
|
| 52
| | Sitio de muestreo
| Indica un dispositivo médico o aplicación de procesamiento de sangre que incluye un sistema dedicado a la recolección de muestras de una sustancia determinada almacenada en el dispositivo médico o contenedor de sangre. NOTA: esto no debe asociarse con un sitio en un paciente donde se toman muestras. |
| 53
| | Camino fluido
| Indica la presencia de una vía de fluido. NOTA: el término "fluido" significa líquido o gas. |
| 54
| | No pirogénico
| Indica un dispositivo médico que no es pirogénico. |
| 55
| | Gotas por mililitro
| Indica el número de gotas por mililitro. NOTA: se especifica el número de gotas por mililitro; tal como 20 y será reemplazado por el número apropiado de gotas por mililitro (ml o mL). |
| 56
| | Filtro líquido con tamaño de
poro
| Indica un sistema de infusión o transfusión del dispositivo médico que contiene un filtro de un tamaño de poro nominal particular. NOTA: se especifica el tamaño de poro nominal del filtro; 15 se muestra como ejemplo y será reemplazado por el tamaño de poro apropiado. |
| 57
| | Válvula de una vía
| Indica un dispositivo médico con una válvula que permite el flujo en una sola dirección. NOTA: es importante que el usuario sepa que el flujo solo es posible en una dirección y no se puede invertir. |
| 58
| | Autopruebas
| Indica que es un agente de diagnóstico in vitro para autopruebas. |
| 59
| | No para autopruebas
| Indica que el dispositivo (se aplica solo a las pruebas rápidas) no está diseñado para autodiagnóstico. Es una prueba rápida que solo debe ser utilizada por un médico capacitado o un profesional de laboratorio en una forma apropiada. |
| 60
| | Pruebas de diagnóstico
ambulatorias
| Indica que el dispositivo solo debe ser utilizado cuando el paciente está cerca de un profesional de la salud. Por lo general, tales pruebas se utilizan en ambulancias, unidades de emergencia, hogares de pacientes o lugares de trabajo, etc. |
| 61
| | Prueba rápida, destinada para
uso exclusivo en el laboratorio
| Indica que es una prueba rápida, destinada para uso exclusivo en el laboratorio y no está diseñada para que la realice el paciente, por lo que solo debe ser realizada por un médico capacitado o un profesional de laboratorio en una forma apropiada. |
| 62
| | Número de paciente
| Indica un número único asociado con un paciente individual. NOTA 1: la marca almohadilla (#) es parte del símbolo. El número del paciente aparece junto al símbolo. NOTA 2: el uso sería para indicar un campo de entrada de datos o una ubicación (por ejemplo, pantalla de entrada del dispositivo médico o tarjeta de implante) o la información proporcionada al paciente. |
| 63
| | Nombre del paciente
| Indica el nombre del paciente. NOTA: el uso sería para indicar un campo de entrada de datos (por ejemplo, pantalla de entrada de dispositivo médico o tarjeta de implante) o en información proporcionada al paciente. |
| 64
| | Identificación del paciente
| Indica los datos en la identificación del paciente. NOTA: el uso sería para indicar un campo de entrada de datos o una ubicación (por ejemplo, pantalla de entrada de un dispositivo médico o tarjeta de implante) o en la información proporcionada al paciente. |
| 65
| | Sitio web de información del
paciente
| Indica un sitio web donde un profesional de salud puede obtener información adicional sobre el producto médico. Este símbolo irá acompañado de la información del sitio web (URL), será adyacente a símbolo. NOTA: el uso es para indicar la información proporcionada al profesional de salud. |
| 66
| | Centro de salud o médico
| Indicar la dirección del centro de salud o del médico donde se encuentra la información médica del paciente. Este símbolo irá acompañado, de la dirección del centro asistencial o del médico junto a éste. NOTA: el uso es para indicar un campo de entrada de datos o una ubicación (por ejemplo, pantalla de entrada de un dispositivo médico o tarjeta de implante) o en la información proporcionada al paciente. |
| 67
| | Fecha
| Para identificar la fecha en que se ingresó la información o se realizó un procedimiento médico. NOTA: el uso es para indicar un campo de entrada de datos o una ubicación (tal como, pantalla de entrada del dispositivo médico o tarjeta de implante) o en la información proporcionada al paciente. |
| 68
| | Dispositivo médico
| Indica que el artículo es un dispositivo médico. |
| 69
| | Traducción
| Para identificar que la información del dispositivo médico original ha sido objeto de una traducción que complementa o reemplaza la información original. Este símbolo irá acompañado del nombre y la dirección de la entidad responsable de la traducción, será adyacente al símbolo. NOTA: si varios de los símbolos (es decir, fabricante legal, fabricante, importador, distribuidor, acondicionador) identifican a la misma entidad responsable, no es necesario duplicar el nombre y la dirección. Este símbolo solo debe usarse cuando la actividad de traducción fue realizada por alguien que no sea el fabricante. |
| 70
| | Reacondicionado
| Para identificar que se ha producido una modificación en la configuración del envase del dispositivo médico original. Este símbolo estará acompañado del nombre y la dirección de la entidad responsable de la actividad de reacondicionado, junto al símbolo. NOTA 1: puede ser necesaria información adicional (es decir, fecha de reacondicionado). NOTA 2: si hay varios símbolos (es decir, fabricante legal, fabricante, importador, distribuidor, acondicionador) que identifican a la misma entidad responsable, no es necesario duplicar el nombre y la dirección. Este símbolo solo debe usarse cuando la actividad de reacondicionado fue realizada por alguien que no sea el fabricante. |
TABLA A.2 - Símbolos de seguridad
| No.
| Símbolo
| Clave/función
|
| 1
| | P031 No usar el dispositivo en una bañera, ducha o depósito lleno de agua |
| 2
| | P031 No alterar el estado del interruptor |
| 3
| | P070 Prohibir meter el dedo en la boquilla de un Hidromasaje |
| 4
| | Advertencia; Material radiactivo o radiación ionizante |
| 5
| | Riesgos biológicos |
| 6
| | Advertencia; Rayo láser |
| 7
| | Advertencia; Radiación no ionizante |
| 8
| | Advertencia; Electricidad |
| 9
| | Peligro Material tóxico |
| 10
| | Advertencia; elemento punzante |
| 11
| | Advertencia; Sustancia corrosiva |
| 12
| | Advertencia; Radiación óptica |
| 13
| | Advertencia; Sustancia o mezcla que representa un peligro a la salud |
________________________________